Expression of His tagged proteins in E. Coli followed by purification using nickel agarose beads is the method of choice to produce most proteins at Solidzymes. This is probably the most common method to produce recombinant proteins at lab scale, however, each lab does it a little differently. In this post we describe purification of a SUMO tagged glucose-6-phosphate dehydrogenase (SUMO-G6PDH) at Solidzymes and characterize its activity under various conditions.
As part of the pentose phosphate pathway G6PDH converts glucose-6-phosphate into 6-phosphogluconolactone while reducing NAD+ to NADH. It is often used as a reporter enzyme in coupled enzyme assays because of the change in absorbance at 340 nm that comes from NADH. In our case, we are interested in coupling this enzyme to a thermostable NADH oxidase. Accordingly, we chose a thermostable G6PDH that has been previously cloned and characterized (Li et al. 2016). A His tag and SUMO leader sequence were added to the N-terminal to facilitate production of the enzyme without any extra amino acids in its final form (Butt et al. 2005). We expect to use this enzyme as part of a co-immobilized enzyme cascade. Therefore, conditions for activity characterization were selected based on compatibility with enzyme carriers and NADH oxidase.
Materials and Methods
Codon optimized DNA corresponding to the sequence of a glucose-6-phosphate dehydrogenase from Thermoanaerobacter tengcongensis with an appended N-terminal 6X His tag and SUMO tag (Figure 1) was synthesized at Genscript Inc. The sequence was cloned into a pET28a plasmid while maintaining the start codon at its correct place downstream of the ribosome binding site. The plasmid was transformed into ECOS Sonic E. Coli cells (a BL21 DE3 derivative) using calcium chloride to render the cells competent. Cells containing the plasmid were identified on agarose plates containing 50 ug / mL kanamycin. Five individual colonies were collected from the plate and grown overnight in Luria-Bertani (LB) media with 50 ug / mL kanamycin. Part of the cell culture was frozen with glycerol for storage in -80 C. The remainder was induced using IPTG and the expression from each colony / culture was evaluated using SDS-PAGE. Once the highest expresser was identified the other cell stocks were discarded.

Figure 1. Amino acid sequence of His-SUMO-G6PDH construct. Purple = 6X His tag with linkers, Orange = SUMO leader sequence, Black = G6PDH.
The frozen cell stock was used to inoculate a 15 mL culture of LB + kanamycin which was grown overnight. The next morning this seed culture was used to inoculate larger shake flasks containing LB media with kanamycin (1.5 L total). These were allowed to grow at 37oC until reaching OD600 of 2.0, then 0.5 mM IPTG was added to induce expression and refrigeration was started to drop the temperature to 30oC. The cells continued to grow over four hours to a final OD of 4.0. Then they were harvested by centrifugation. Cells were resuspended in a minimal volume of Resuspension Buffer (20 mM HEPES pH 7.5, 150 mM NaCl, 1 mM DTT) and frozen for later use.
Frozen cells were thawed and sonicated for 14 minutes on ice to lyse the cells. Cell debris was pelleted by centrifuging for 20 minutes at 18,000 xg. The supernatant was removed and mixed with 1 mL of Ni-NTA for 30 minutes to bind His tagged proteins. Then the mixture was transferred to an Econocolumn (BioRad) for efficient nickel agarose washing. The sample was washed ten times with Wash Buffer (20 mM HEPES 7.5, 150 mM NaCl, 1 mM DTT) then eluted with 1 mL fractions of Elution Buffer (20 mM HEPES, 150 mM NaCl, 1 mM DTT, 250 mM imidazole, pH 7.5) (Figure 2). The concentration of G6PDH in each fraction was quantified by measuring the absorbance at 280 nm on a Nandrop One spectrophotometer (blanked with elution buffer). The total yield after purification was 9.2 mg or 6.2 mg / L of shake flask culture.
The imidazole was removed by dialysis in 3 L of stirred Wash Buffer at 4oC. The SUMO tag was removed by incubating the protein with SUMO protease (also purified on site) at a 1:100 protease:G6PDH ratio for 1 day at 4oC (Figure 2). After tag cleavage the SUMO tag and protease were removed from solution by binding to 0.5 mL of nickel agarose. Finally, the purified and processed G6PDH samples were frozen in -80oC with 10% glycerol added. The purity of the sample was estimated at 95% using gel densitometry (SDS-PAGE and ImageJ analysis).

Figure 2. Purification of 6X His tagged SUMO-G6PDH fusion enzyme. (a) Elution profile of ten fractions collected from nickel agarose. (b) SDS-PAGE of SUMO-G6PDH before, during, and after tag removal by SUMO protease.
Results
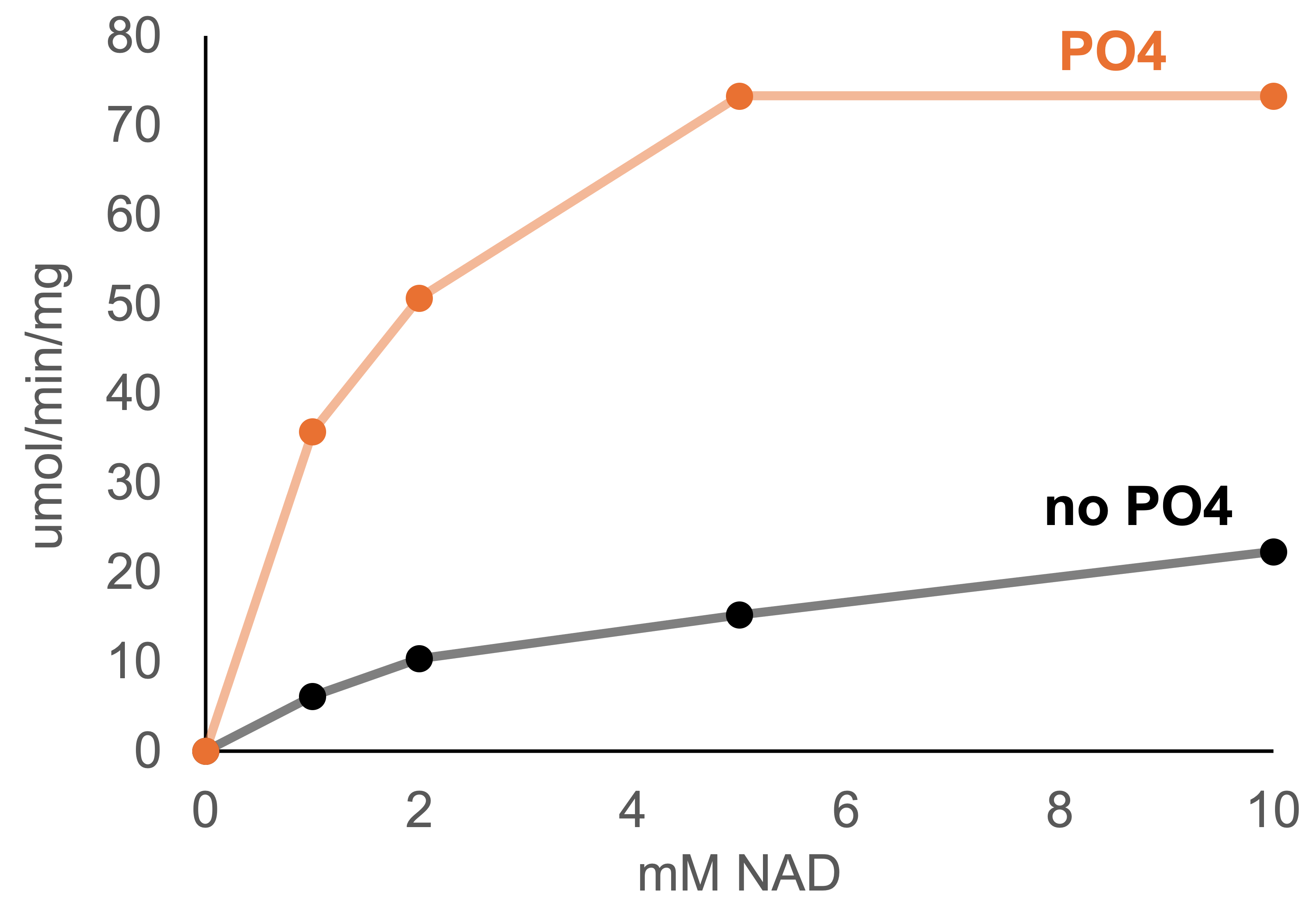
G6PDH reduces NAD+ to NADH while oxidizing glucose-6-phosphate. This particular enzyme prefers to use NAD(P)+ as a cofactor, but we used NAD because this substrate is preferred by the NADH oxidase it will be paired with in the future. An activity assay was developed following the absorbance of NADH at 340 nm in a Biotek Synergy H4 Hybrid platereader. The enzyme was activated by phosphate and performed best at pH 7.0 (Figure 3). Interestingly, 100 mM phosphate shifted the Km from 3.6 mM NAD+ (consistent with the available literature) down to 1.7 mM. Thus, including a high concentration of phosphate is a useful approach to conserve relatively expensive NAD+.

Figure 3. Conditions for optimal G6PDH activity. (a) pH series in HEPES buffer (b) The result of buffering with HEPES (black) versus phosphate (orange). The pH ranged from 6.8 – 7.5 during NAD+ titration (due to acidity of NAD+).
The activity of G6PDH was regularly measured at 60oC. However, to see the effect of different temperatures activity assays were also conducted at 50, 40, and 30oC. Additionally, G6PDH was divided into parts and incubated at 50, 60, or 70oC prior to activity measurements in order to evaluate the stability of the enzyme over time (Figure 4). The half-life was approximately 5, 10, and 20 hrs at 70oC, 60oC, and 50oC. This suggests that the highest yield over time might be found in the 40 – 50oC range, but this requires more detailed experimentation.

Figure 4. The effect of temperature on G6PDH activity and stability. (a) The activity at different temperatures in a temperature-controlled platereader. (b) The remaining activity after incubating G6PDH at 50, 60, and 70oC.
Conclusion
This enzyme was successfully purified and stored for future coupled enzyme assays with NADH oxidase. The information gathered on buffer conditions and temperature will be needed to plan these future experiments. We are optimistic that it will be a useful tool for characterizing NADH oxidase activity in soluble form, as well as in immobilized form. Purification of this enzyme with the SUMO tag and subsequent removal of the tag with SUMO protease also validated the SUMO system for protein expression for the first time at Solidzymes. This system will be used in the future to facilitate purification and promote solubility of challenging proteins.